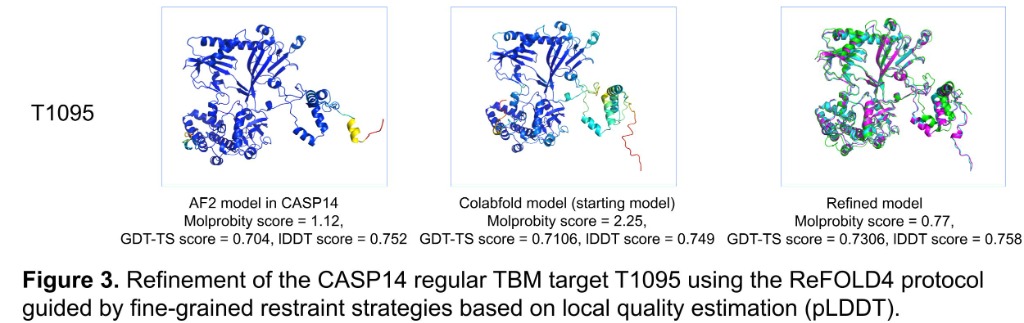
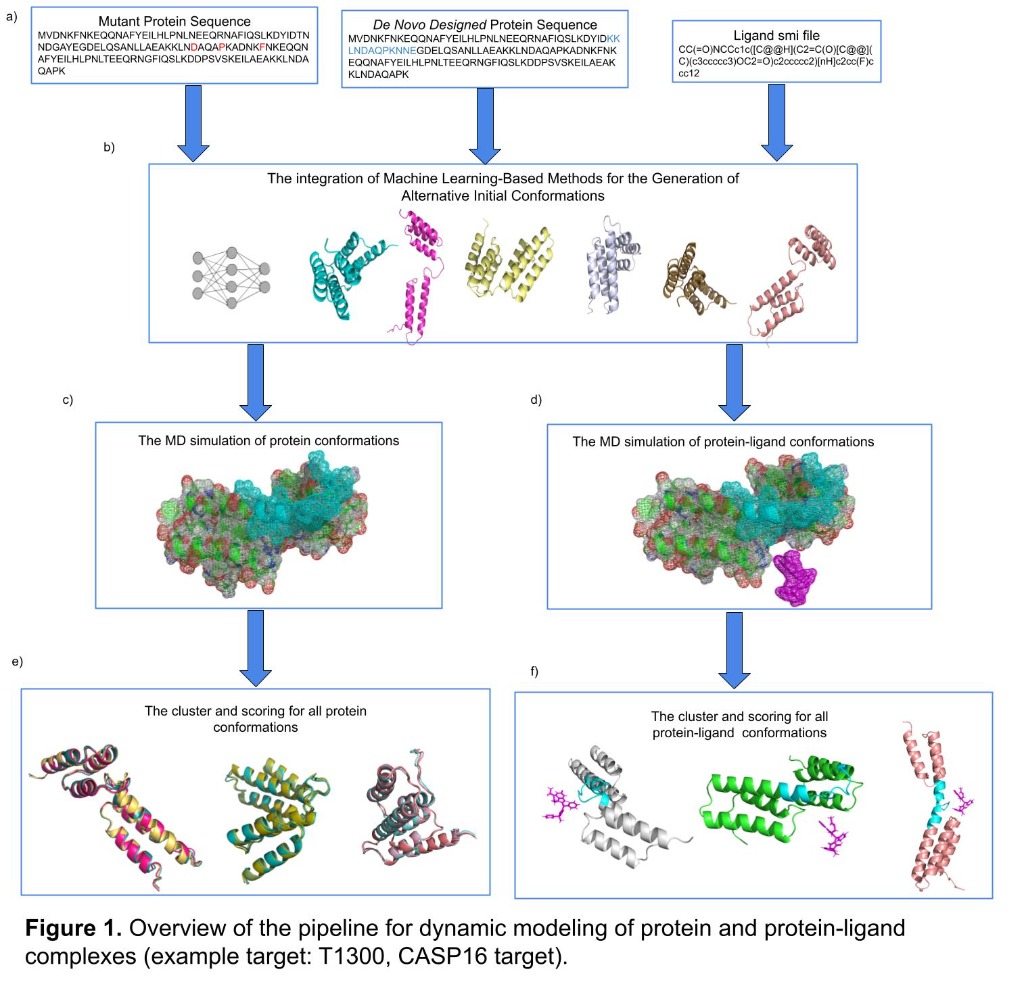
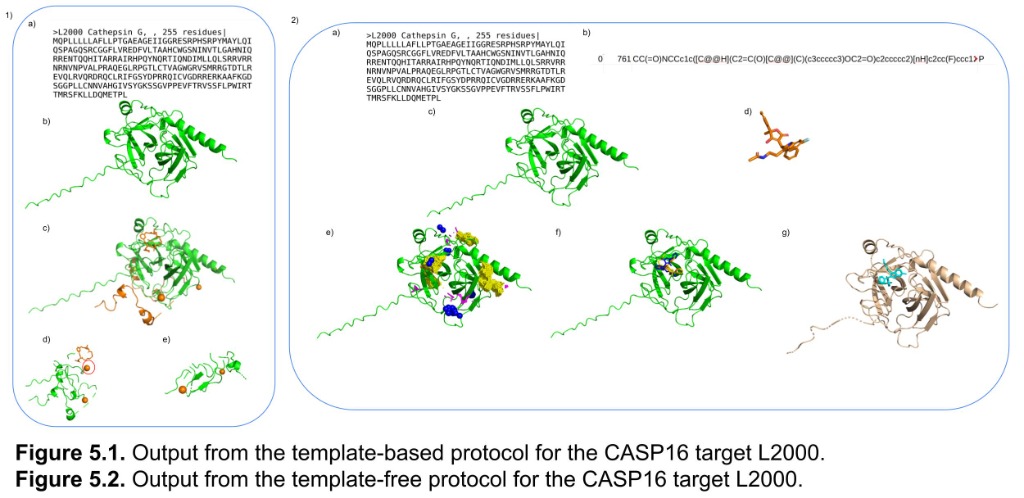
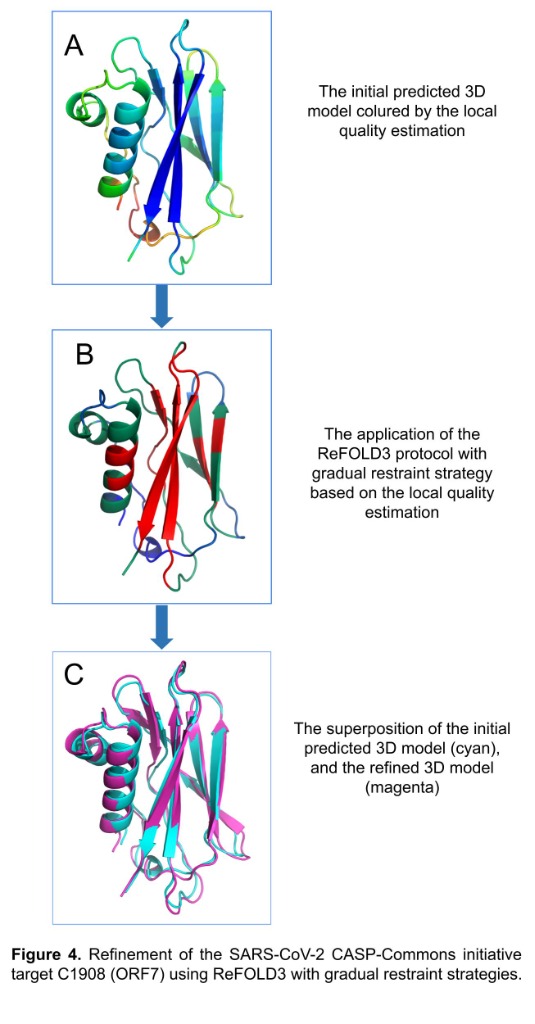
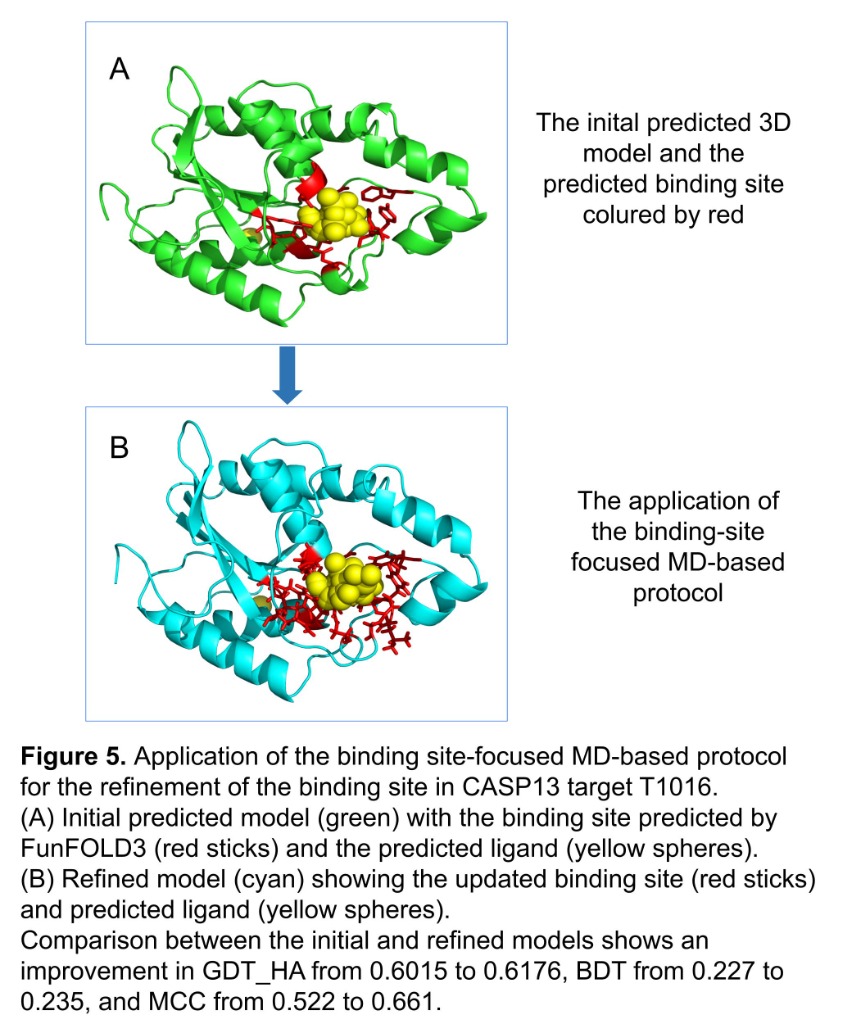
Computational structural biologist with 9+ years of research experience in protein structure prediction, molecular dynamics simulation, and AI/ML-driven drug design. Developer of multiple internationally benchmarked tools (IntFOLD, ReFOLD, FunFOLD, MultiFOLD, ModFOLDdock2Q) that consistently rank among the top methods at CASP blind prediction experiments. Published 15 peer-reviewed papers (H-index 9, 530+ citations) in journals including Nucleic Acids Research, Blood, and Bioinformatics Advances. Currently combining deep learning structure predictors (AlphaFold2/3, Boltz-2, Chai-1) with physics-based methods for cancer vaccine design in collaboration with InstaDeep/BioNTech. Experienced in Python, PyTorch, HPC/SLURM environments, and end-to-end bioinformatics pipeline development.